
Case Report | DOI: https://doi.org/10.58489/2836-5062/016
*Corresponding Author: Kavita.V
Citation: Kavita.V, Kavitha.A, Shreya Kar. (2024). Real-World Performance of Ai-Powered Ich Triage System. Journal of Clinical Oncology Reports. 3(1); DOI: 10.58489/2836-5062/016
Copyright: © 2024 Kavita. V, this is an open-access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 23 January 2024 | Accepted: 02 February 2024 | Published: 16 February 2024
Keywords: Sacral Chordoma, Sacrococcygeal, primary brain tumours
Chordomas are rare malignant tumours arising from primitive notochordal remnants and comprises of 0.2 % of primary brain tumours and less than 5 % of primary bone tumours [1]. Only 5 % of them occur in the first two decades. Only 300 paediatric cases have been reported so far in the literature [2-5]. The average age at diagnosis in children is 10 years with a male-to-female ratio being close to 1. Adult chordomas are primarily found in the sacro-coccygeal region whereas the majority of paediatric chordomas are intracranial. Metastatic spread seems to be more common in children under 5-year-old with more frequent in case of sacro-coccygeal locations and undifferentiated histology. The tumour location majorly determines the clinical presentation. Sacrococcygeal forms may present with an ulcerated subcutaneous mass, radicular pain, bladder and bowel dysfunctions. Diagnosis is suspected on computerised tomography showing the bone destruction and with typically lobulated appearance, hyperintense on T2-weighted magnetic resonance imaging. Management of these tumours is based on possible complete primary resection followed by local irradiation, ideally proton beam therapy. A major prognostic factor identified in chordomas was age of onset and it is notable that the worst progression occurs in very young children, under the age of 5 years. Prognosis is better in children than in adults except for the aggressive form of chordomas occurring in children under 5 years of age. The brachyury growth factor (growth factor T) is a specific marker of chordomas, implicated in notochordal development. Its locus 6q27 is frequently amplified within chordoma cells and its inactivation can block growth of chordoma tumour cell lines (U-CH1) in vitro
Chordomas are rare malignant tumours arising from primitive notochordal remnants and comprises of 0.2 % of primary brain tumours and less than 5 % of primary bone tumours [1]. Only 5 % of them occur in the first two decades. Only 300 paediatric cases have been reported so far in the literature [2-5]. The average age at diagnosis in children is 10 years with a male-to-female ratio being close to 1. Adult chordomas are primarily found in the sacro-coccygeal region whereas the majority of paediatric chordomas are intracranial. Metastatic spread seems to be more common in children under 5-year-old with more frequent in case of sacro-coccygeal locations and undifferentiated histology. The tumour location majorly determines the clinical presentation. Sacrococcygeal forms may present with an ulcerated subcutaneous mass, radicular pain, bladder and bowel dysfunctions. Diagnosis is suspected on computerised tomography showing the bone destruction and with typically lobulated appearance, hyperintense on T2-weighted magnetic resonance imaging. Management of these tumours is based on possible complete primary resection followed by local irradiation, ideally proton beam therapy. A major prognostic factor identified in chordomas was age of onset and it is notable that the worst progression occurs in very young children, under the age of 5 years. Prognosis is better in children than in adults except for the aggressive form of chordomas occurring in children under 5 years of age. The brachyury growth factor (growth factor T) is a specific marker of chordomas, implicated in notochordal development. Its locus 6q27 is frequently amplified within chordoma cells and its inactivation can block growth of chordoma tumour cell lines (U-CH1) in vitro
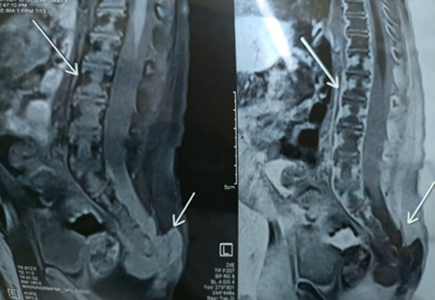
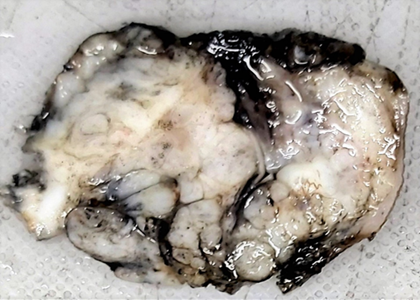
A 1-year-old male child presented with a swelling in the sacral region for 1 year. For last 4 months he developed bladder and bowel dysfunction. MRI whole spine revealed focal lytic lesion and destruction involving distal sacrococcygeal region with adjacent T2 hyperintense presacral, posteriorsacral and lateral soft tissue component. Epidural extension of the soft tissue was visualised. (Fig 1) Presacral component is seen abutting the lower rectum. Radiology imaging findings were suspicious of sacrococcygeal teratoma. Wide excision of the swelling was done and sent for histopathology. Grossly, a grey tan to yellowish nodular tissue measuring 5.2 X 4 X 4 cm was received in the department of histopathology, Metropolis Healthcare Limited. Cut section showed a grey tan firm lesion measuring 5.0 X 3.8 X 3.7 cm which was 0.2 cm from inked circumferential margin (Fig 2). Microscopically, the sections revealed tumour composed of lobules and sheets of epithelioid cells with abundant clear to eosinophilic cytoplasm displaying bubbly and vacuolated appearance. Immunohistochemistry was done and it was positive for EMA and S-100(Fig 3a, 3b). After confirmation of the diagnosis, the patient has been referred to the oncologist for further treatment.


Chordomas are primary bone tumors, which arise from notochord remnants along the axial skeleton. It is a slow growing tumour, which is locally invasive and destructive, eventually leading to compression of surrounding structures including vessels and nerves. Symptoms depend on the site involved and size of the tumour. Intracranial tumors can produce headache, defects in the visual field, dysphagia, cranial neuropathies and radiculopathies; cranial nerve VI is commonly affected. Sacral location can result in back pain, dysfunction of sphincter, radiculopathies, and motor weakness of lower extremities.
Diagnosis begins with radiological imaging and MRI is considered superior to other modalities where lesion becomes hypodense on T1 flair.
Histology confirms the diagnosis. There are 3 histological subtypes: conventional, dedifferentiated and poorly differentiated. Conventional type is the classical one composed of large epithelioid cells with clear to eosinophilic bubbly cytoplasm (physaliphorous cells) separated in lobules by fibrous septa. Chondroid chordoma is a subtype in which large area of matrix mimicking hyaline cartilage are present. In immunohistochemistry, these tumours are diffusely immunoreactive for S-100, EMA and cytokeratin with diagnostic hallmark being expression of brachyury. Dedifferentiated chordomas are biphasic in appearance characterized by conventional chordoma and high-grade sarcoma. The two components are usually abruptly separated. In immunohistochemistry, the dedifferentiated component can show focal cytokeratin expression but does not express brachyury. Poorly differentiated neoplasm with notochordal differentiation usually arising in the axial skeleton are characterized by loss of SMARCB1 expression. Microscopically, the physaliphorous cells with classic features and extracellular myxoid stroma are absent. Geographical necrosis is conspicuous. These tumours are immunoreactive for CK, S-100, brachyury with diagnostic feature being loss of SMARCB1 (INI1) expression.
Differential diagnosis includes both benign and malignant entities. Benign entities include Ecchordosis physaliphora (EP) and benign notochordal cell tumour (BNCT). Both these entities contain cells with abundant vacuolated clear to eosinophilic cytoplasm and immunoprofile is identical to chordoma. The differentiating factors are: well delineated borders, lack lobular architecture, necrosis, conspicuous mitosis and high-grade nuclei. BNCT also lacks extracellular myxoid matrix. Malignant entities includes chondrosarcoma which may have similar radiological features as chondroid chordoma but is negative for epithelial markers- CK, EMA and brachyury; myoepithelial tumours of soft tissue or bone which shows similar histomorphological and immunohistochemical features except for negative brachyury expression; myxopapillary ependymoma and metastatic carcinoma. The treatment consists of complete surgical resection with negative margins followed by high dose, high precision radiation therapy in few cases. The extent of surgical resection plays a major role in determining the length of the disease-free interval (6,7).
Chordomas are malignant neoplasms with notochordal differentiation that primarily involve the axial skeleton, often making complete surgical resection difficult or impossible. Treatment options have largely been centered on surgical excision with negative margins, however recently identified molecular alterations have led to ongoing clinical trials using targeted therapy.