Case Report | DOI: https://doi.org/10.58489/2836-8657/011
*Corresponding Author: Gonzalez-Torres Jorge
Citation: Gonzalez-Torres Jorge, Gutierrez-Rodriguez Georgina, Garcia-Hidalgo Linda, Corona-Herrera Judith, Montante-Montes Daniel, Dominguez-Cherit Judith. (2024). Merkel cell carcinoma, a rare sarcoma. Journal of Surgery and Postoperative Care. 3(1). DOI: 10.58489/2836-8657/011
Copyright: © 2024 Gonzalez-Torres Jorge, this is an open-access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 17 August 2023 | Accepted: 16 January 2024 | Published: 07 March 2024
Keywords: Merkel cell carcinoma, cutaneous neoplasm
Merkel cell carcinoma (MCC) is a rare cutaneous neoplasm that accounts for less than 1% of all primary cutaneous neoplasms.1 A trabecular carcinoma was first described by Toker in 1972. It was named Merkel cell because it expresses Merkel cell markers, which are still being discussed. Regional incidences range from 0.1 to 0.88 / 100,000 person-years, but they are higher in countries like New Zealand and Australia.2 The majority of MCC cases (80%) are caused by Merkel Cell Polyomavirus (MCPyV) clonal integration into the host genome. In the remaining 20% of cases, UV mutations are responsible, and prolonged UV exposure is a factor.3,4 MCPyV early regions contain large and small T antigens, which have been shown to drive tumorigenesis. These proteins may play a role in MCC as well. Cells express a truncated form of LT protein that cannot replicate the virus but retains the domain that inhibits Retinoblastoma (Rb), the tumour suppressor.
Merkel cell carcinoma (MCC) is a rare cutaneous neoplasm that accounts for less than 1% of all primary cutaneous neoplasms.1 A trabecular carcinoma was first described by Toker in 1972. It was named Merkel cell because it expresses Merkel cell markers, which are still being discussed. Regional incidences range from 0.1 to 0.88 / 100,000 person-years, but they are higher in countries like New Zealand and Australia.2 The majority of MCC cases (80%) are caused by Merkel Cell Polyomavirus (MCPyV) clonal integration into the host genome. In the remaining 20% of cases, UV mutations are responsible, and prolonged UV exposure is a factor.3,4 MCPyV early regions contain large and small T antigens, which have been shown to drive tumorigenesis. These proteins may play a role in MCC as well. Cells express a truncated form of LT protein that cannot replicate the virus but retains the domain that inhibits Retinoblastoma (Rb), the tumour suppressor. MCC without MCPyV exhibit cellular genomic mutations in tumour suppressor genes, especially TP53 and RB1. Other tumour suppressors, including NOTCH genes and KMT2D, are inactivated less frequently.5 Based on a systematic review conducted in Japan, the following clinical characteristics were found: male: female ratio 1:1.6, mean age 77.5 years; 63.0% of the tumours were found in the head and neck, 5.2% in the trunk, 12.6% in the upper limbs, 15.1% in the lower limbs, 3.5% in the buttocks, and 0.6% in the genitals; mean tumour size was 2.79 cm, MCPyV detected in 68.9%, immunosuppression noted in 6.8%.6 We present two cases of this rare tumour that are larger than usually reported in the literature and have rare topography; one is immunosuppressed, and the other is sun exposed.
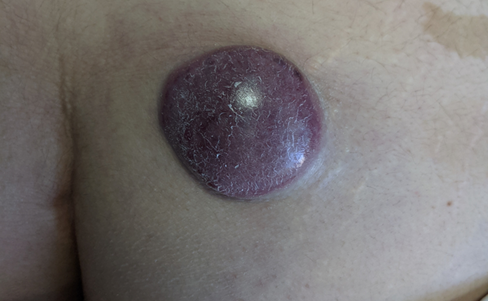
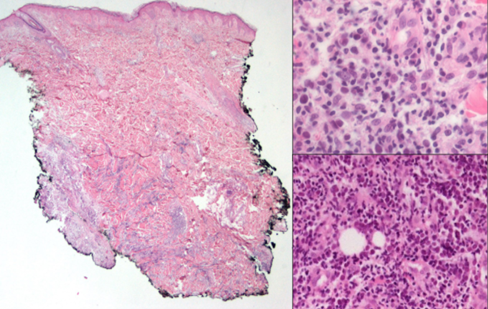
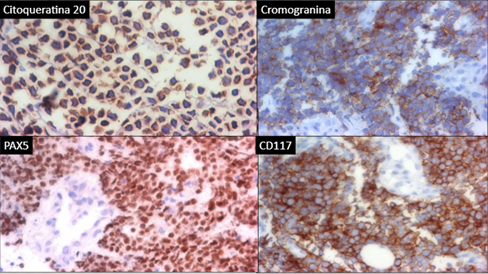
This woman, 48 years old and a stay-at-home mom, was diagnosed with low-risk hairy cell leukaemia in 2019 and was given cladribine to treat it. In 2020, she was seen by the dermatology team and diagnosed with a dermatosis with a neoformation. It was in her right gluteus and looked cupuliform with a diameter of 4 cm. The surface was smooth and shiny, with purple coloration, and the edges were regular and smooth, with a solid consistency. When it was looked at through a microscope, there were unstructured, white, and pink areas (picture 3A). She had 3 months of growth and was growing rapidly, but she wasn't feeling well. A spindle biopsy was performed, the pathology report was invasive MCC, with deep dermis invasion CK-7, CK20 +, Chromogranin +, Synaptophysin -, Ki67 80%, CD56 +, CD117 +, it was negative for TTF1 and SV40. Subsequently, the oncology service classified the patient as an IV EC patient, and he was subsequently treated with with surgery and etoposide and cisplatin chemotherapy.
In 2021, an elderly man, a flea market vendor, and a type 2 diabetic had a dermatosis with exophytic necrolysis, which was located on the right upper limb, affecting the anterior side of the arm, distal third, and elbow. The necrolysis had a diameter of 8x12 cm, was multilobladed in appearance, and had a reddish-purple coloration. The surface of the necrolysis was ulcerated and friable, and it was mobile to the touch. Dermoscopy revealed an unstructured area of white and pink (Figure 3B). The patient had a history of more than one year of painful, intermittently bleeding, limited limb mobility, and continued growth without medical attention. The patient underwent a spindle biopsy, which resulted in a diagnosis of soft tissue carcinoma. The pathology report revealed that the cancer was Merkel cell carcinoma CK20+, with a paranuclear pattern and synaptophysin+ and chromogranin+; it was free of TTF1. Subsequently, the oncology service classified the patient as an IV EC patient, and he was subsequently treated with nivollumab.
The clinical presentation of the two cases highlights the wide variation in topographic and morphological presentation, making it a diagnostic challenge, and, as with all neoplasms, the diagnosis is histological; they are predominantly located in the Head and Neck, and were found in the Upper extremity and Buttock, the latter being the second most common images reported in the literature.6,9 The morphology of the lesion is consistent with that found in other publications. The subcutaneous necro-formations are red to vultureous, well-circled, and may include ulceration and pedunculation.8,9 The dermoscopic features of the lesion were linear irregular, polymorphous, poorly focused, milky-pink, white, and structureless. Architectural disorder was present, but pigmented structures were not present in all lesions.9 In the two cases, the lesion had structureless white or pink-red areas. Among the histological differential diagnoses are osteosarcoma, rhabdomyosarcoma, desmoplastic small cell tumour, small cell or amelanotic melanoma, mesenchymal chondrosarcoma, Ewing sarcoma, lymphomas, and neuroblastoma. De metastatic small cell lung cancer is frequently the most challenging distinction.11,12 A majority of MCC express cytokeratins, CK20 in 95% of cases, neuroendocrine markers (synaptophysin, chromogranin, and CD56), and neurofilament (NF). Thyroid transcription factor-1 (TTF-1) and CDX-2 are negative. CK7 is generally negative, but occasional reported cases are positive.12 In the present case, a combination of neural filaments, chronic lymphocytic cells (CK-20), chronic lymphoblastic cells (CK7), and thyroid transcriptase factor-1 (THF-1) stains has a high degree of sensitivity and specificity to differentiate MCC from histopathological mimics.7 For treatment of localized disease, resection of the tumour with wide margins is recommended, with secondary closure plus radiotherapy. If lymph node involvement, resection of it plus radiotherapy. In metastatic disease or recurrence, the treatment of choice includes immunotherapy with anti-PD1 and anti-PDL-1. Avelumab became the first FDA-approved checkpoint inhibitor for MCC, as a first-line treatment, had an ORR of 62.1% in patients with stage IV disease, Nivolumab (anti-PD-1) administered 240 mg every 2 weeks as a first-line or second-line treatment exhibited tumour regression response in patients with metastatic MCC in phase I/II clinical trial, ORR was 68% with 82% progression-free survival and 92% overall survival at 3 months.13,14,15 Only one case was indicated for anti-PD-1 treatment due to the high cost of treatment, making it only available to a limited number of patients. About the forecast MCC primary tumour size cutoff of 2 cm is still considered a significant breakpoint in prognosis 7, in both cases the dimensions were larger, which is a negative prognostic factor. The relative 5-year survival in distant cases 19%. Related factors are nodal involvement, the presence of metastases, and a size greater than 5 cm.16.
Clinical Diagnosis of Merkel Cell Carcinoma is a challenging and uncommon neoplasm requiring a high level of suspicion. In the present case report, two patients have been diagnosed with this neoplasm. These cases illustrate the entire spectrum of the disease. Both neoformations are associated with ultraviolet radiation exposure and immunosuppression respectively. However, the presentation size of these neoformations exceeds the reported literature size, indicating a poor prognosis. This is likely due to the delays in medical care caused by the ongoing SARS-CoV-2 pandemic. New treatments are available for advanced stages of the disease; however, the cost of these treatments renders them out of reach in our country.